Viral infections continue to appear in human and wildlife populations, causing frequent epidemics and occasional pandemics. The Section of Genomic Epidemiology and Evolution of Pathogens (GEEP) at DIEPS, FIC, NIH aims to answer critical questions about pathogen emergence and establishment in animal and human populations using evolutionary and epidemiological information extracted from viral genomes. Specifically, our Evolutionary and Computational Virology project seeks to unravel viral host origins, the molecular signatures that promote pathogen transmission between hosts, the mechanisms of pathogen evolution within hosts, and the dynamics of spatiotemporal spread. We utilize integrated molecular biology, computational, and Bayesian statistical approaches to uncover the viral phylodynamic processes and better inform public health responses.
Current projects include investigations into the origins, evolution, and epidemiology of influenza A virus, seasonal coronaviruses, SARS-CoV-2, RSV, HIV, and rabies virus.
Projects
Origins and Evolution of Seasonal Human Coronaviruses
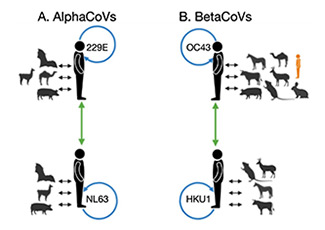
The 229E, NL63, OC43, and HKU1 seasonal human coronaviruses (sHCoVs) are endemic globally, however, little is known about their evolutionary history. Using a multigene and complete genomes analysis approach, we find the evolutionary histories of sHCoVs to be more complex than previously recognized, owing to frequent recombination of CoVs, including within and between sHCoVs. Depending on the gene studied, we observed a variety of potential ancestors for OC43 and 229E. HKU1 comprised two genetically divergent genotypes (A and B), possibly representing two independent transmission events from murine CoVs, with genotype B being genetically more diverse than all the other sHCoVs.
The Evolutionary History and Spatiotemporal Patterns of Respiratory Syncytial Virus
Phylodynamic analysis of the local and global patterns of spread of the respiratory syncytial virus (RSV) and identification of the factors affecting the spatial dynamics at different scales.
COVID-19 Breakthrough Infections in Vaccinated Health Care Workers
The evolutionary dynamics of SARS-CoV-2 breakthrough infections in Mexican healthcare workers and patients immunized with various vaccines. Molecular evolutionary analysis of 400+ SARS-CoV-2 sequences from Yucatan, Mexico sequenced in partnership with the Autonomous University of Yucatan and the COVID-19 International Research Team (COV-IRT) partners at the New York Genomics Center (NYGC).
The Early Dynamics of SARS-CoV-2 in Côte d’Ivoire - An Epidemic Seeded by Travel Fluxes
Investigate SARS-CoV-2 transmission and evolution in the early stages of the COVID-19 pandemic in Côte d’Ivoire, using viral genomes collected from patients with detailed travel history information. We aim to reconstruct the routes and timeliness of viral introductions into the country and assess the relative contribution of each introduction in terms of how the epidemic evolved over time.
Publications
-
Global dissemination of influenza A virus is driven by wild bird migration through arctic and subarctic zones, co-authored by Fogarty's Nídia S Trovão
Molecular Ecology, October 14, 2022 -
COVID-19 Exposure Assessment Tool (CEAT): Exposure quantification based on ventilation, infection prevalence, group characteristics, and behavior, co-authored by Fogarty's Nídia S Trovão
Science Advances, September 30, 2022 -
Origins and Evolution of Seasonal Human Coronaviruses co-authored by Fogarty’s James Otieno, Joshua Cherry, David Spiro, and, Nídia Trovão
Viruses, July 15, 2022 -
Ecological divergence of wild birds drives avian influenza spillover and global spread, featuring Fogarty's Nídia S Trovão as co-author
PLoS Pathogens, May 19, 2022 -
A comprehensive SARS-CoV-2 and COVID-19 review, Part 1: Intracellular overdrive for SARS-CoV-2 infection, featuring Fogarty's Nídia S Trovão as co-author
European Journal of Human Genetics, May 16, 2022 -
The emergence and transmission dynamics of HIV-1 CRF07_BC in Mainland China, featuring Fogarty's Nídia S Trovão as co-author
Virus Evolution, February 17, 2022 -
SARS-CoV-2 introductions and early dynamics of the epidemic in Portugal, featuring Fogarty's Nídia S Trovão as co-author
Communications Medicine, January 28, 2022 -
Proposal for Human Respiratory Syncytial Virus Nomenclature below the Species Level, featuring Fogarty's Nídia S Trovão as co-author
Emerging Infectious Diseases, June 27, 2021 -
Genomic diversity of SARS-CoV-2 during early introduction into the Baltimore-Washington metropolitan area, featuring Fogarty's Nídia S Trovão as co-author
JCI Insight, March 22, 2021
Contact
Members
- Nídia S. Trovão
- Joshua L. Cherry
- Sana Tamim
- Erin Yuan