NIH Websites Are Changing
NIH is moving the launch of its new website to the fall to ensure the best possible experience for everyone who relies on our websites for health information, research, funding opportunities, and other resources.
Get the Latest Updates and FAQs
Research needed to treat sickle cell disease in Africa
November / December 2014 | Volume 13, Issue 6
By Cathy Kristiansen
Most children with sickle cell disease in Africa suffer relentless pain, experience numerous infections and die before their fifth birthday. Although treatments exist, they are not widely available in sub-Saharan Africa, where 70 percent of the world's children with sickle cell disease are born, according to the WHO. NIH is funding a number of efforts to develop low-cost methods to diagnose the condition and investigate affordable treatments to help reduce the suffering.

Photo by Micah Albert, courtesy of Photoshare
Three-quarters of children who inherit sickle cell disease
are born in sub-Saharan Africa, but most have no access
to treatment and die before their fifth birthday.
Most of the interventions used today for sickle cell disease were produced for wealthy countries with higher health budgets and patients already protected by better nutrition and vaccines against infectious diseases.
"Supporting sickle cell research in sub-Saharan Africa and other low-resource settings can make a difference," said Dr. George Mensah, who focuses on global health research at NIH's National Heart, Lung and Blood Institute. "We cannot be paralyzed by the enormity of the challenges."
A global movement is growing to address the inequity in sickle cell disease treatment. The WHO named it a public health priority in 2006, followed by the UN in 2008. "There's a total sea change in interest and what's being done for sickle cell disease patients and the understanding of our moral and scientific relationship with them," said Fogarty grantee Dr. Richard S. Cooper, of Loyola University in Chicago. "Investors and scientific institutions have begun to take the issue much more seriously," he added.
Sickle cell causes pain, early death
Sickle cell disease, one of the world's most common genetic disorders, occurs when a child inherits a trait from each parent that causes most of their red blood cells to form into crescents, rather than discs. Affected blood is less able to carry oxygen and flow smoothly, which causes a host of health problems and a shorter lifespan. Children who inherit only one trait are protected against severe malaria, which is why the disease is most prevalent in malaria endemic parts of the ancient world, primarily Africa, the Middle East and Southeast Asia. Countries in Equatorial Africa bear the greatest burden. (See "Burden of Sickle Cell Disease" chart below.)
With population migration, the disease has spread widely, including to the U.S., where the disease affects roughly 100,000 people, according to the CDC.
Sickle cells were only discovered in human blood in 1910, but the hereditary disease evolved in humans at least 5,000 years ago because of the survival advantage it gives against malaria. Just how this protection works is not fully understood, but studies suggest the sickle cell trait alters biochemical and immune mechanisms in the blood, compromising the malaria parasites' ability to grow and mature.
The NIH has supported research into the condition since 1942 and currently funds nearly 140 grants to study the disease and devise ways to treat its symptoms and complications. Standard approaches in high-income countries include strong pain relief medications, the drug hydroxyurea and blood transfusions. Several preventive steps are also available, such as genetic testing and counseling, assisted reproductive technology, and vaccines and prophylactic antibiotics to avert infections. Most patients receiving these interventions reach their 40s or 50s, before they face potentially fatal conditions like pulmonary hypertension and organ failure.
Without treatment, the disease takes its toll early, as children are battered by chronic pain, anemia, repeated infections, stroke, leg ulcers or breathing difficulties. This affects not only the child, but the entire family of caretakers, bringing major social and economic consequences, including missed school and work days and income loss.
Budget-constrained health authorities in Africa commonly fund treatment of infectious diseases that threaten high death tolls, such as malaria, tuberculosis and HIV/AIDS, or those covered by childhood vaccinations, rather than relatively rare conditions that are expensive to diagnose and treat. At most, some patients with sickle cell disease might receive a handful of vaccinations, antibiotics when bacterial infections erupt, medication for severe pain and, especially if they supply money and suitable donors, blood transfusions to lessen physical crises.
Research needed to inform policy decisions
Researchers are working to change that, however, with several studies currently underway in developing countries aimed at providing sound evidence to convince policymakers of the feasibility and benefits of offering programs to diagnose and treat sickle cell disease. Scientists agree that children with a double trait derive clear advantages from the pneumococcal vaccine - not universally administered in sub-Saharan Africa - and protective antibiotics during the early years of life. The decision to invest in hydroxyurea is less clear-cut.
Two key projects were recently launched in Nigeria and Uganda to study the safety and effectiveness of the drug, the main intervention used in wealthy countries. Hydroxyurea, designed as a cancer treatment, has proved remarkably effective at spurring production of normal-shaped fetal hemoglobin, which relieves symptoms in sickle cell disease. It was approved in 1998 to treat adults in the U.S., more recently for use in children. A daily pill, mixed into food if need be, it markedly cuts the frequency of severe pain episodes, hospitalizations, lung damage and blood transfusions.

Photo by Liz Gilbert,
courtesy of Photoshare
Researchers in Uganda and Nigeria are
testing a drug that alleviates sickle cell
disease symptoms - but can also depress
the immune system - to ensure it is safe
for children in malaria zones.
But the drug's main side effect is to depress infection-fighting white blood cell counts - a matter of particular relevance in Africa, with its heavy burden of immune system challenges from pathogens and malnutrition. For instance, studies have shown that children with two sickle cell traits are more vulnerable to death from malaria than other infected children, so it is important to gauge if hydroxyurea's immune system depression adds to this risk in malaria-endemic areas.
"In some places, the position of the government and the experts has been that hydroxyurea should not be used in Africa," according to Cooper. "There has been great concern that you would expose these children to a higher risk from infection." One of the scientists Cooper trained with support from his Fogarty International Research Collaboration Award is leading the drug study in Nigeria. Dr. Bamidele Tayo, a Loyola University investigator, is conducting a pilot to examine whether a low, fixed dose of the drug will benefit patients without also harming them. The project is funded by the Doris Duke Charitable Foundation.
To raise the dose to higher levels would require frequent monitoring, which is unrealistic given the setting. Before achieving widespread use, it's important to determine the trade-off between maximum efficacy and what strapped health systems might be convinced to provide, Cooper said.
The drug costs about $1 per day in the U.S. and half that for its cheapest generic form available elsewhere. "It's an old drug, off patent," Cooper noted. "But even 50 cents a day can be too much for a family in Africa."
Meanwhile, a Ugandan study is looking specifically at hydroxyurea's interplay with malaria. Researchers recently launched a phase 3 clinical trial of the drug in children aged one to four, who live in a malaria-prone area of Uganda - where sickle cell trait prevalence reaches 45 percent in some pockets. The NOHARM study is led by Dr. Chandy C. John of the University of Minnesota and Dr. Christopher Ndugwa of Makerere University. Funded by the Doris Duke Charitable Foundation, its goal is to examine whether the drug elevates the risk of fatal complications from malaria. "It's possible that kids on hydroxyurea, once they get malaria, could be at higher risk for more severe malaria, but we are hoping to show otherwise," John explained. Participants in Fogarty's Global Health Program for Fellows and Scholars have helped to get the study up and running. (See the related article,
Fogarty Fellow Dr. Juliana Anyanwu studies sickle cell drug in Uganda.)
The Ugandan health ministry is very interested in the study's outcome, John noted. If hydroxyurea is determined to be both safe and effective, it might become part of routine care for all children with sickle cell disease in the country. "Identifying young children early, before the disease has had a chance to damage their organs, and treating them early is the key to a better life for these kids," John said.
Include in HIV treatment platform
Existing health systems developed to diagnose and treat HIV/AIDS, as part of the U.S. President's Emergency Plan for AIDS Relief, could easily be expanded to also check infant blood samples for sickle cell disease. "I think the time has come to think of how you bundle and leverage resources to maximize the benefits from infrastructure that's already there," Mensah observed.

Photo by Arturo Sanabria,
courtesy of Photoshare
Existing HIV health platforms and lessons
learned in other low-resource countries
could be used to improve the lives of
people with sickle cell disease in Africa.
However, there would have to be a commitment to also provide some level of treatment, said Dr. Julie Makani of Muhimbili University and the Wellcome Trust, who is collaborating with NIH. "Ethically, you can't diagnose them and say, 'Sorry, we have nothing for you.' You do need a level of service to be present before you can screen."
That's why researchers like Cooper and John are working to produce data they hope will demonstrate that the existing interventions for sickle cell disease can bring clear benefits for sub-Saharan Africa. Positive results may also motivate stakeholders to negotiate affordable pricing for the region so that diagnostics, vaccines and drugs can be added to existing patient services.
Even if treatment were inexpensive and widely available, more research would be required, according to Mensah, who asked, "Would it be acceptable to the community? Do they know enough about the benefits and the harm? And what have we done in terms of providing health education and communication to make sure there's acceptance of the effective interventions?" The NIH can play an important role in helping to answer these and other scientific questions, he said.
Lessons learned in Jamaica
Sickle cell disease has been studied in Jamaica for more than 40 years and has produced some findings that may apply in Africa. Funded by the Wellcome Trust and the UK Medical Research Council, the work was led for more than three decades by Dr. Graham Serjeant. A high proportion of Jamaicans have African ancestry; about 10 percent carry the sickle cell trait and one of every 300 children born has the burden of two traits.
In a recent talk at NIH, Serjeant shared some lessons learned in Jamaica. As in other low-resource settings, pneumococcal infection puts Jamaican children at great risk, he said. Penicillin prophylaxis offers some protection, but patients often have difficulty taking the medication consistently. Researchers tested whether a monthly injection would encourage more compliance than pill-taking, and they found that not only did it reach almost 90 percent, but it was also associated with less invasive pneumococcal disease. Another study showed additional protection is provided from the pneumococcal vaccine - providing policymakers with evidence they could use in deciding how to allocate funding for the greatest impact.
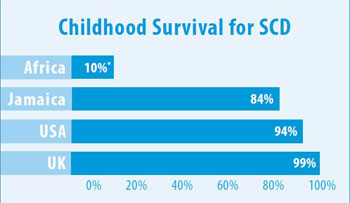
*Note on Africa 10%: Survival for most African children without access
to treatment is estimated at 10 percent. For those living in urban areas
and whose families are able to purchase care, survival rates may be
closer to 50 percent.
See
chart sources below.
Another intervention studied in Jamaica concerns acute spleen sequestration, in which sickled red blood cells become trapped in the spleen, enlarging it and causing a life-threatening emergency. Researchers taught caretakers how to palpate a child's spleen, recognize when it is swollen and immediately seek help at a clinic. "The message is, you can teach your mothers to diagnose this complication," Serjeant said. "In Jamaica the death rate from this cause dropped from 28 percent to 3 percent."
Cooper has been establishing a research collaboration among his Loyola colleagues, Jamaican scientists and their Nigerian counterparts to build capacity for genetic and clinical studies of sickle cell disease at the University College Hospital in Ibadan. With Fogarty support, the investigators have held training sessions, and compared data and lessons learned to enhance patient care in Nigeria. The South-South collaboration provides an excellent opportunity to transfer advances in clinical care that are appropriate for low-resource settings, Cooper said.
Jamaica currently only has the resources to check about 40 percent of its newborns for sickle cell disease, but it is working to expand screening to cover all babies. It is trying to reduce the number born with the disease, by offering genotyping to 16,000 high school students so they can discover if they have the trait before they select a mate.
It's too early to know the outcome but researchers hope it will have an impact. Sickle cell trait screening has reportedly been very successful in Saudi Arabia and Bahrain but may be difficult to replicate in settings with different cultural practices and levels of health resources.
Genomics may hold key to cure
Genomics is another area that holds promise for advancements in sickle cell disease. The
Human Heredity and Health in Africa initiative, known as H3Africa, was recently established by the NIH and Wellcome Trust to train a cadre of African geneticists and establish genomic biorepositories on the continent.
H3Africa recently convened a gathering of sickle cell disease experts in Tanzania, including scientists, clinicians, policymakers, educators and others. They discussed the accumulated global research, considered how to build more evidence and capacity, and agreed to form an information-sharing network.
Sickle cell disease is caused by a single genetic mutation, yet patients can have mild, moderate or severe forms of the disease. Studies could focus on both the genetic and environmental factors. Among other urgent research priorities is to investigate the genetic variety of double sickle cell trait carriers. About 5 percent of them show few symptoms throughout their lives. By studying them, scientists may discover a clue that could lead to new treatments or even a cure.
For now, policymakers need to reassess how resources are allocated in Africa with a focus on reducing preventable deaths and disabilities, maintains the NIH's Mensah. "When children don't have the opportunity to make it to age 5, that should be a national priority."
More Information
Access a collection of resources and background information on sickle cell disease from NIH and more.
- NIH sickle cell news and resources
- NHLBI topic:
Sickle cell disease
-
Sickle cell disease drug proves safe, effective in African children
NHLBI/NIH news, December 1, 2018 - NHLBI Publication:
Sickle Cell Disease: Milestones in Research and Clinical Progress, 2018
-
Specific dosage of sickle cell drug increases survival rate
NHLBI/NIH news, November 17, 2015 -
Sickle cell anemia treatment so successful in kids that trial is halted
NHLBI/NIH news, November 19, 2014 - NHLBI Publication:
Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, 2014
-
Sickle cell disease
Genetics Home Reference, National Library of Medicine (NLM) -
Hydroxyurea information
MedlinePlus, National Library of Medicine (NLM)
- Related NIH webcasts
- Other sickle cell resources
- Additional background resources
Chart Sources and Descriptions
- Source for the chart, "Burden of Sickle Cell Disease":
- Sources and description for the chart, "Childhood Survival for SCD":
-
Sickle Cell Disease in Africa: A neglected cause of early childhood mortality. Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN.
American Journal of Preventive Medicine, December 2011.
-
Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study. Klaas JJ Wierenga, Ian R Hambleton, Norma A Lewis, Sickle Cell Unit, et al.
Lancet, Volume 357, Issue 9257, March 2001
-
Survival of children with sickle cell disease. Quinn CT, Rogers ZR, Buchanan GR.
Blood, June 2004.
-
Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Telfer P, Coen P, Chakravorty S, Wilkey O, Evans J, Newell H, et al.
Haematologica, July 2007.
- Description of "Childhood Survival for SCD" chart showing burden of sickle cell births per year by country: Nigeria 91,011; Dem. Rep. Congo 39,743; Tanzania 11,877; Uganda 10,877; Angola 9,017; Cameroon 7,172; Zambia 6,039; Ghana 5,815; Guinea 5,402; Niger 5,310; Sub-Saharan Africa Total 242,187; Worldwide Total 305,773.
To view Adobe PDF files,
download current, free accessible plug-ins from Adobe's website.